原标题:科普 | 什么是Alport综合征(Alport syndrome,AS)
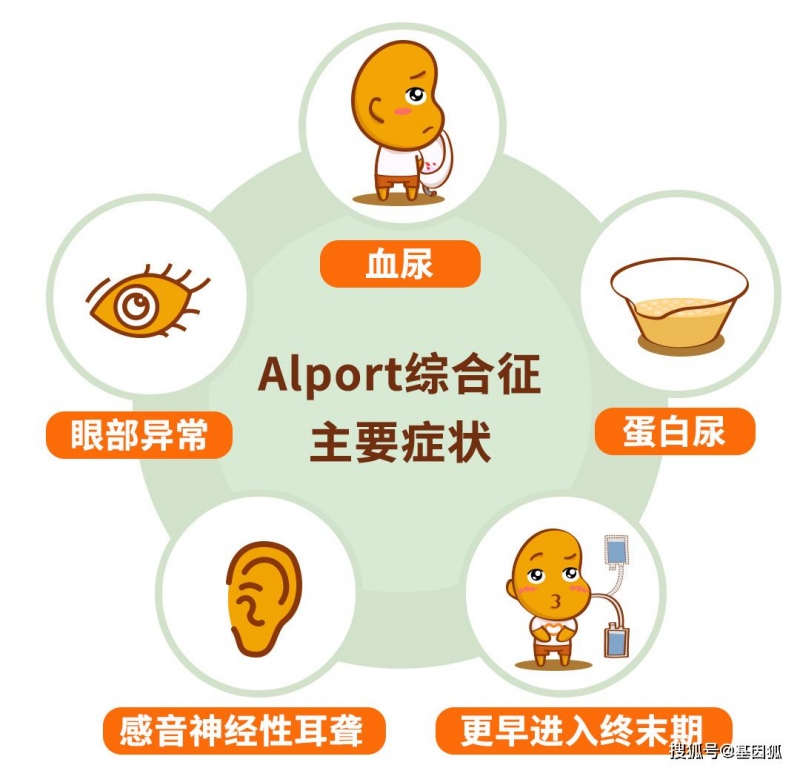
Alport综合征(Alport syndrome,AS)亦称遗传性进行性肾炎,是原发性肾小球基底膜疾病,该病由编码基底膜Ⅳ型胶原蛋白α3~α5链的基因COL4An(n=3,4,5)骤变所造成的。Alport综合征疾病的命名至今已有80年,期间阅历了临床症状的描绘、对肾脏病理尤其是肾小球基底膜超微结构的知道、相关致病基因的发现和确认等几个重要的里程碑性的开展。临床特点是血尿、蛋白尿及进行性肾功用减退,部分患者可兼并感音神经性耳聋、眼部反常、食管滑润肌瘤等肾外体现。
为何需求确诊Alport综合征?
临床上AS并非稀有。据文献报导AS在肾小球疾病中约占2%,在终晚期肾衰病人中约占5%,在肾移植病例中约占2.3%,在成人肾活检中占0.3%,而在儿童肾活检中约占1.7%-2.5%。一起AS具有临床异质性,临床体现多样。
现在临床上针对AS并无特异性的医治办法,但研讨显现经过早确诊早干涉对症医治可有用推迟患者抵达终晚期肾脏病的时刻。Gross等(2012年)研讨显现ACEi前期干涉Alport综合征患者,可推迟AS患者抵达ESRD的时刻达13年。
别的,AS具有遗传性,其遗传办法可掩盖三种经典的孟德尔遗传规则,包含伴X染色体显性遗传、常染色体显性遗传、常染色体隐性遗传。清晰AS致病基因的骤变位点及遗传办法,可经过遗传咨询、无创产前筛查或植入前AS基因检测,完成优生优育。
最终,对开展到ESRD需进行肾脏代替医治时,对供肾者挑选和移植肾功用前期监测也有含义。若挑选亲属供肾,杂合的COL4A5基因骤变女人携带者如患者的母亲,若临床体现无蛋白尿、肾功用减退和耳聋,可当作供肾者,而男性不能作为供肾者,因为他们或许处于肾脏疾病的开展期。因为部分AS患者在肾脏移植后会发作抗肾小球基底膜(GBM)抗体,发作移植肾抗GBM肾炎,因而AS患者在术后一年内需亲近重视血清抗GBM抗体及肾功用。
临床确诊办法
Alport综合症相关基因
Alport综合症有8个致病基因,包含COL4A3,COL4A4,COL4A5,COL4A6,CD151,FN1,LMX1B和MYH9基因。经过检测基因的骤变方式(包含单碱基骤变,刺进/缺失和染色体拷贝数变异)能轻松完成对AS的检测。
Alport综合征患者肾功用逐步损失。简直一切患者的尿液中都有血液(血尿),这表明肾脏功用反常。许多 Alport综合征患者的尿液中也含有很多的蛋白质(蛋白尿)。跟着病况的开展,肾脏的功用越来越差,导致终晚期肾病(ESRD)。
Alport综合征患者在幼年晚期或青少年前期常常会呈现由内耳反常引起的感音神经性听力损失。受影响的人的眼睛或许有变形的晶状体(前小扁豆)和眼睛后部感光安排(视网膜)的不正常色彩。这些眼睛变形很少导致视力下降。
优生优育
对有生育需求的Alport综合征患者或家系成员,攻略主张其在怀孕前进行基因检测,确认基因骤变的位点,并由具有临床遗传学常识的医师进行合理的遗传咨询和生育辅导。一起,已孕妈妈主张做产前基因确诊,以完成优生优育。
以上内容仅供参考,不能代替医师和其他医务人员的主张、确诊、医治及用药根据,如有身体不适,请及时就医。做Alport综合征基因检测的费用请咨询基因狐。
参考文献
Alport综合征确诊和医治专家引荐定见,[J].中华肾脏病杂志, 2018, 34(3): 227-231
责任编辑:
